If we reduce wastage, drug prices won’t go down
Posted: May 30, 2016 Filed under: General industry topics Leave a commentBack in March, the NY Times published an article claiming that we lose $3 billion dollars each year because of the cancer drug that are wasted. The article was inspired by a paper from Memorial Sloan Kettering Cancer Center which came up with the daunting $3 billion number. The crux of the argument is that drug companies sell their products in packages that leave physicians no choice but to throw out the “extra” drug that remains after a dose is administered. If drug companies simply sold their products in more convenient sizes, we could reduce this waste and recoup some of that $3 billion.
Going even further than that, Peter Bach, one of the authors of the paper, infers that drug companies are intentionally doing this to boost their profits! That’s quite an accusation.
The leftover drug still has to be paid for, even when discarded, making it possible for drug companies to artificially increase the amount of drug they sell per treated patient by increasing the amount in each single dose vial relative to the typically required dose. – Peter Bach
In reality, reducing the wastage of drugs would have no impact whatsoever on the price of drugs. And more importantly, if drug manufacturers followed Peter’s advice and came up with more vial sizes to reduce waste, that would be going against FDA guidance and likely result in an increase of mis-dosing of cancer drugs.
It is true that cancer drugs are wasted. In fact, it’s practically unavoidable for certain drugs that are dosed based on body-weight. Since patients come in all shapes and sizes, you would need an infinite number of vial sizes in order to make sure no wastage occurred. But the question is, would reducing wastage reduce drug prices?
The answer to the question is no. Drug companies determine price independent of packaging or vial size. Let’s look at a hypothetical drug, Product A. If Product A is 10% better than a drug on the market, you might chose to price it at a 10% premium (yes, it’s more complex than that, but bear with me). If the competing drug is $1000/month, you would then price Product A at $1100/month. Keep in mind you haven’t even thought about your packaging or dosing at this point. Pretty straightforward so far.
Let’s look at a couple of extreme examples to see how you might price Product A under different vial size scenarios: (1) only one large vial size is produced and (2) 5 smaller vial sizes are produced. How would each one be priced?
Option 1: One large vial (200 mg)
This vial would be large enough so that any patient would require only one vial. The math is easy on this one, just price the vial for an average patient ($1100/month). Yes, for smaller patients, most of the vial would be thrown away, but every single patient would cost $1100/month.
Option 2: Five smaller vials (1 mg, 5 mg, 10 mg, 25 mg, 50 mg)
Since there are so many more vial sizes, there will be a lot less wastage in this scenario. Before we look at costs, lets do some math so we have the numbers we need.
If the average patient should cost $1100/month the the price per mg is $11/mg ($1100/month for average patient who take 100 mg per month).
If an average patient weights 70 kg and is taking 100 mg per month, the dose is 1.4 mg/kg.
Looking at the costs across three different patient types:
Small patient (50 kg) – they need 70 mg per month (1 x 50mg and 2 x 10mg vials), so they cost $770
Average patient (70 kg) – they need 100 mg per month (2 x 50mg vials), so they cost $1100/month
Large patient (90 kg) – they need 129 mg per month (2 x 50mg, 1 x 25mg and 4 x 1mg vials, $1414/month
What do you end up with if you average costs across all patients? A cost of $1100/month. Nothing has changed except you wasted a lot less of the drug and the large patient’s physician or nurse needs to gather up 7 vials in 3 different sizes and make sure there are no mistakes!
Now you might be saying “but you’re not wasting as much drug! Isn’t that a positive thing?” Yes, reducing waste would reduce manufacturing costs, so yes, it is a positive thing in isolation. But what if we look at the big picture?
First off, raw material costs are a very small part of the cost for most pharmaceuticals. The cost of goods sold (COGS) for most drugs is less than 10% of the price and often only a few percent. However, if you chose to create more vial sizes, that has additional expense as well. You have more vials to manufacturer, more validation studies to do, etc. Overall, the cost of having more vials might be more than the savings on raw materials!
However, the most important reason to limit the number of vial sizes is patient safety, as show in the “large patient” example above. That’s why the FDA prefers fewer rather than more vial sizes. If you go in for a chemotherapy session, do you know if the pharmacist grabbed the right vial? You do if there is only one size available. Fewer vial sizes means less of an opportunity for dosing mistakes.
In conclusion, the paper published by Memorial Sloan Kettering has good intentions, but missed out on the bigger picture. Reducing drug waste would likely result in no cost savings whatsoever and an increase in medical errors.
An unprecedented looked at how biotech prices their drugs
Posted: February 21, 2016 Filed under: Latest news Leave a commentWhether you’re in the biopharma industry or not, you’ve heard about the controversy surrounding the pricing of Sovaldi and Harvoni, two therapies for hepatitis C that have revolutionized the treatment of a very common and sometimes deadly disease. The topic first came up when Sovaldi was launched December 2013, but the noise still hasn’t died down thanks to people like Martin Shkreli and companies like Valeant.
At the same time there have been a lot of views expressed about how drugs companies price drugs and how they should price drugs. The interesting part is that very few people know how drug pricing is done today. That shouldn’t be surprising since those types of decisions are highly confidential and no company is interested in releasing any information to the public.

That all changed last December when the Senate Finance Committee released a report on their investigation into the pricing of Sovaldi and Harvoni. You can download the entire, 144-page report here. When I started to go through it, my jaw dropped. This report provides an incredible in-depth look at how Gilead priced both drugs, including: (1) market research results from physicians and payers, (2) minutes from closed-door meetings among Gilead executives that led to the final pricing strategy and (3) post-launch responses from private and public payers.
If you are at all interested in this topic, I suggest you spend a couple hours reading through the report. I will warn you though, I thought the report did show some bias in supporting the government’s position that Gilead acted recklessly. Regardless, it is an unprecedented look at how biopharma companies price their drugs.
FDA dismisses study that shows generic drugs are lower quality than branded version
Posted: April 10, 2014 Filed under: Science 2 CommentsIn this blog post I’m going to take a step away from the business side of the pharmaceutical industry and dive a little bit deeper in to the science.
In March of last year, a paper (paywall) and a poster were published that claimed generic versions of Pfizer’s Lipitor (atorvastatin) had quality problems that may impact public health. The study in question was run by a physician at Brigham and Women’s hospital in Boston by the name of Preston Mason. I don’t know about you, but a study from a very well respected institution such as Brigham and Women’s gets my attention, especially if it concerns drug quality. If you take a closer look at the results, they are quite alarming. The study found that every single generic drug they tested had very high levels of an impurity, sometimes up to 30%! This is a important issue in the industry right now as several foreign generic manufacturers, such as Ranbaxy, have been placed under FDA bans that prevent their drugs from being imported to the United States.

This is quite a shocking result as the FDA standards that exist rarely allow more than a few tenths of a percent of impurities in a final product. To find impurity levels as high as 30% would suggest very lax quality standards at these generic manufacturers and would warrant an immediate recall and possible sanctions for the manufacturer.
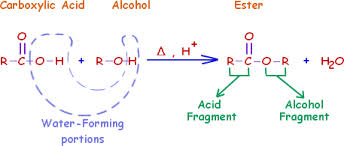
If we dive into the science a little deeper, we see that the impurity that was found was the methyl ester of Lipitor. For those who are not chemists, esters are very simple derivatives of carboxylic acids that are formed from the acid and an alcohol. The diagram below shows how the alcohol is incorporated into the molecule.
At first glance the analytical methods used in the paper seem quite robust. The scientists obtained pure reference samples of both atorvastatin (the active molecule in Lipitor) and the methyl ester impurity. The researchers even went so far as to test the biologic activity of impurity to show that such high levels of the impurity would reduce the effectiveness of the drug. However, if we look a little closer, we find a critical error in the analysis that raises some doubt about the findings.
If you examine how the analysis was done, the generic drug samples were prepared for analysis by dissolving them in methanol. This is a pretty typical method to prepare samples, however in this case, this method may have resulted in formation of the impurity itself. If you look at the ester chemistry diagram above, you can see that if methanol reacts with a carboxylic acid, it forms a methyl ester, the impurity in question.
However, the authors of this paper did run a control. They took some of the atorvastatin reference material and placed it in methanol for 10 weeks and they did see methyl ester impurity forming, however it was a very slow process (it took 10 weeks to convert 20% of the drug to the ester). Apparently their argument is that since it took 10 weeks to form the methyl ester (and they conducted the analysis of the generic drugs much more quickly than that) that the generic drugs must have had that impurity present from the start.
That is where the experimental error was made. The assumption the scientists made is that formation of the methyl ester always happens slowly. However, they only tested this on the pure atorvastatin reference material. The generic drugs that they analyzed contain more than just the active drug, they also contain other compounds used to form the tablet called excipients.
Excipients are a class of chemicals that have many uses in tablet manufacturing. They are used as fillers, binders (make sure the tablet doesn’t crumble), coloring and flavoring agents. Excipient selection is very important as you need to ensure that the drug is stable, but that it also dissolves reproducibly when a patient swallows it. The excipients themselves can have a number of different properties, some are basic (alkaline) and some are acidic. If you look at the ester chemistry diagram above, you’ll see the symbol H+, which represents an acid. The formation of esters is catalyzed by acids. What likely happened in this research was that the excipients in the generic drug catalyzed the formation of the methyl ester “impurity” such that after only a few minutes in methanol a large amount of the methyl ester was formed.
How could these researchers test that theory? Simply use a different alcohol in the preparation of the generic drug samples. If they used an alcohol like ethanol (ethyl alcohol), they very likely would have seen an ethyl ester impurity instead of the methyl ester, which would prove that the “impurity” was being formed by their analytic method, not by the manufacturer of the drug.
This paper is a great example of why the design of scientific experiments is so critical, especially the use of controls. The only way you can prove that something like an impurity is there is by proving you’re not putting it there. This research didn’t do that and that might explain why the FDA just recently dismissed all of the paper’s findings.
How the government makes healthcare more expensive by cutting costs
Posted: January 3, 2014 Filed under: Strategy Leave a commentIncreasing healthcare costs are one of the most important issues facing the United States today. Although the rate of increase has slowed down over the past few years, the amount of money spent on healthcare in the US has risen at an alarming rate over the past decade. It’s not only an issue for private insurers as the impact of Medicare and Medicaid spending also threatens to balloon the federal budget. Incredibly, cost savings measures instituted by the government have, and will, lead to further increases in healthcare costs. How will this happen? Lets focus on the treatment of cancer for a moment; the cause and effect should be clear.
When a cancer patient requires treatment such as chemotherapy, they are typically treated in one of two settings: (1) a community-based oncology clinic or (2) a hospital-affiliated clinic. Community-based clinics are private clinics owned by the doctors who practice there and can vary in size from only a few physicians to 50 or more. These clinics are run like small businesses with physicians paying themselves a salary and the clinic taking either a profit or loss at the end of the year. Hospital-affiliated clinics look very similar to community clinics from the outside, but differ in that the hospital system handles all of the accounting and the physicians who work there are typically paid a salary.
Another, very important difference between the two settings is that treating a cancer patient in a hospital-affiliated clinic is typically much more expensive than treating a patient in a community clinic. Data gathered by Avalere shows that, on average, the cost of treating a cancer patient is 20% to 55% more in a hospital-based setting (table 3 in this report).

Now, if you wanted to reduce the cost of healthcare, it would seem prudent to encourage cancer patients to receive their care at community-based clinics. If you’re saving roughly a third per patient on average, it adds up to a significant amount of money. The reality is that the government has instituted two different programs that are pushing patients away from community-based clinics towards hospital-affiliated clinics. And incredibly, both of these programs were instituted to reduce healthcare costs.
The first government program was an attempt to reduce the amount spent on the drugs used to treat cancer. Typically, oncologists are reimbursed through “buy-and-bill.” Physicians purchase a cancer drug using their own money and once they have used it to treat a patient, they bill the patient’s insurance company. Insurers, both public and private, typically pay physicians more than what the drug actually costs in order to cover some of the overhead associated with treating a patient. This extra money is typically referred to as the “spread.” In the past, the spread was quite generous; if a doctor bought a drug for $10,000, they might get an additional 10 or 20% ($11,000 or $12,000 back from the insurer). As healthcare costs continued to rise, Medicare and Medicaid decided to reduce their reimbursement levels so as to provide as little as an additional 4.2% in spread and there is talk of lowering it further. These changes have drastically reduced the revenue that community clinics take in and in many cases has forced doctors to sell or close their community-based practices. A recent report has shown that in 2012 there was a 20% increase in both oncology clinics closing or merging with existing hospital systems. Between 2005 and 2011, the percentage of cancer patients treated in the hospital setting increased by 150% (from 13.5% to 33%). As the number of community-based clinics decreases, more patients get treated in the more expensive hospital-affiliated clinics.
The other factor driving patients to hospital-affiliated clinics is the 340b program. Initially designed to assist hospitals who treat patients with little or no health insurance, the program allows certified hospitals to purchase out-patient drugs (such as cancer therapies) at a 23.1% discount (same discount Medicaid gets). This certainly helps those hospitals who treat underinsured and uninsured patients, but the program has a catch: 340b hospitals can purchase all of their out-patient drugs through this program, whether they are used for uninsured or insured patients. In addition, hospitals don’t have to pass along the savings to the insurer, they are allowed to pocket the difference. It isn’t unusual for oncology therapies to cost more than $100K/yr per patient, so that 23.1% discount becomes a lot of money, all of which goes to the bottom line of the hospitals. To give you an example, the Duke University Hospital system effectively doubled their profit margin on drugs to 53% for a gross profit of $70M. This has created a huge incentive for hospitals to both become 340b certified and for them to attract oncology patients to their clinics.
The situation we now have is that cancer patients are being pushed out of the less expensive community-based setting as oncologists struggle to stay profitable and pulled into the more expensive hospital-affiliated clinics as hospitals seek to capture as much 340b business as possible. Not exactly a great way to save money now is it?
Controlling healthcare spending will be a priority for the US in the coming decade. In order for that to happen, we need a coordinated effort by the government and private insurers to find a way to incentivize not only quality care but also cost-efficient care. Without an understanding of the economic pressures and incentives offered by the current system, this may prove a very difficult task.
A setback for the hygiene hypothesis
Posted: October 29, 2013 Filed under: Innovation 2 CommentsIt’s always interesting when a clinical trial you’ve been watching reads out. One of the first posts I wrote for this blog was about a clinical trial that Coronado Biosciences was running based on the hygiene hypothesis. The company was testing the efficacy of T. suis ova (pig whipworm eggs) in treating Crohn’s disease.

Well, Coronado Biosciences just released the results of a phase 2 trial (TRUST-1) and they are not positive. It was a double-blinded, placebo-controlled trial of 250 patients with moderate to severe Crohn’s disease, so a positive signal would have been pretty convincing evidence of the efficacy of the therapy. The primary endpoint of the trial measured response using the Crohn’s Disease Activity Index (number of patients who see a >100-point decrease in CDAI) and the secondary endpoint measured induction of remission (number of patients who achieve a CDAI score of less than 150). For both endpoints, there was no significant difference between the treated patient population and the patients who received a placebo. This is not a “we almost made it” result, but rather a “this doesn’t work at all” result.
The company did see a small signal in patients with very high CDAI scores (> 290), but unfortunately it was not statistically significant. Coronado blamed it on a higher than expected placebo response, which may at first glance seem like a cop-out, but is actually quite common in trials for relapsing-remitting inflammatory diseases.
Another European trial is underway (TRUST-2) so Coronado will have a second chance to prove the therapy works, but things don’t look very promising at this point. Crohn’s disease is a pretty tough market to compete in as several effective therapies already exist. Even if TRUST-2 has a positive read-out, it’s unlikely the efficacy would be enough to support commercialization.
Coronado is also running trials of T. suis ova in several other indications including ulcerative colitis, multiple sclerosis, autism and psoriasis. I’ll be following those trials closely as a positive outcome would be an incredible jump forward in our understanding of the immune system and offer new therapies for patients who often have few other options. I’ll keep you posted.
Follow-up: Only a few days after posting this, Coronado Biosciences halted their TRUST-2 trial due to lack of efficacy. Not surprising, but certainly a nail in the coffin for T. suis ova in the treatment of Crohn’s disease. It will be interesting to see how the other indications fare…
Big pharma’s “pay-to-delay” deals: Helping or hurting patients?
Posted: February 4, 2013 Filed under: Strategy Leave a commentA few weeks ago it was announced that Jon Leibowitz has decided to leave his post at the Federal Trade Commission. Jon has been the driving force behind the FTC’s attempt to ban so called “pay-to-delay” deals that have become increasingly common between branded and generic pharmaceutical companies. The FTC claims that these deals are “anti-competitive” and basically amount to collusion between branded and generic companies. According to the FTC, these deals cost the American public $3.5 billion per year in higher drug cost as cheaper generic drugs are delayed from entering the marketplace. Jon’s efforts seem to have paid off as Senator Chuck Grassley recently introduced legislation banning the deals and the Supreme Court will soon decide if the deals are legal.

You might be wondering what these “pay-to-delay” deals are exactly, so lets look at a hypothetical example:
Let’s say a branded pharmaceutical company is selling a patent-protected drug with annual sales of $5 billion per year. The company’s patent on the drug doesn’t expire until 2020 so they will continue to promote and sell the drug until then, upon which time a generic drug company will begin producing and selling a generic version (after gaining approval from the FDA). Generic companies compete with each other to get their generic version approved first, since it comes with a 180 day exclusivity period where no other generic company can sell their version. During the 180 days, the generic company will undercut the price of the branded therapy and grab a large part of the market share. Since the generic drug company only has to do a fraction of the R&D that the branded company did, the 180 day exclusivity is very profitable. After the 180 days are up, any generic drug company can sell their generic and the price falls to the point where there is little profit to be made.
Where thing get complicated is that generic companies often don’t wait until the branded drug’s patent expires. Instead, they will challenge the validity of the patent in court and if successful, they will get the coveted 180 days of exclusivity and can start selling their generic version immediately. As you can imagine, this potential payoff creates a lot of incentive to challenge a patent.
What does the patent holder do when their patent is challenged? Since there are often billions of dollars of profit at stake, they fight it out in court by hiring some very expensive lawyers to argue that their patent is valid . If the generic drug company has already started selling their generic version before the patent issue is settled (a so called “at risk” launch), the branded drug company can sue for damages if the patent is found to be valid. Since the law allows for recovery of triple damages, losing a patent decision is what generic companies fear the most (for a great example of such an outcome, look no further than Pfizer’s request for $2 billion in damages for Teva’s at-risk launch of Protonix).
As you can see, the stakes are very high for both companies if a patent challenge actually ends up being decided by a judge. It’s an all or nothing outcome: one party will win big and one party will lose big. It’s for this reason that many patent challenges lead to out-of-court settlements that include so called “pay-to-delay” deals. In exchange for a payment from the branded drug company (either in the form of cash or other financial incentives) the generic drug company will agree to delay the launch of their generic version. You can think of this as a way to “meet in the middle”. Both companies get something out of the deal and it eliminates the risk of being on the losing end of a winner-takes all outcome.
However, is the FTC’s allegation that pay-to-delay deals delay entry of cheaper generic drugs and hurt the consumer true? No. The launch of the generic drug is only delayed in the sense it would enter the market later than if the generic company succeeded in invalidating the patent. However, the reason why the generic company agrees to the pay-to-delay deal is because it doesn’t believe it will succeed in invalidating the patent. If pay-to-delay deals were banned, the generic company would likely just pack up its bags and head home as the possibility of losing the patent challenge is more risk than they can tolerate. If anything, pay-to-delay deals actually result in a generic drug entering the market sooner than it would have otherwise.
If pay-to-deal deals are banned and branded and generic companies lose the ability to “meet in the middle”, the availability of generic drugs will likely be delayed in many cases, which is exactly what the FTC is trying to avoid. It will remain to be seen if Chuck Grassley’s bill passes into law, but in the mean time, keep an eye out for the Supreme Court decision. The court’s decision will have far reaching consequences for the pharmaceutical industry and in the end the consumer.
Just how much profit is there in a new drug?
Posted: September 13, 2012 Filed under: General industry topics 3 CommentsIt seems like the debate over the cost of patented drugs never ends: patients complain about the ever increasing prices and the drug companies complain about the high costs of R&D. However, one question you rarely see asked is how much does it really cost to make that one dose of a new drug? How much of the price is profit?
Now keep in mind when I say profit I really mean contribution margin. Contribution margin is price minus the variable cost of producing a single unit and ignores the fixed costs which in the case of a new drug includes the cost of R&D, marketing and the equipment used to manufacture the drug, just to name a few. It’s called a contribution margin because it’s the part of the revenue that contributes to paying for the fixed costs. A good example of a contribution margin comes from the entertainment industry: once you’ve shot a film, paid the actors, edited the film and put it on a DVD, what is the cost of producing another copy of the DVD compared to the price it’s sold for?
In the world of pharmaceutical manufacturing, the variable cost of producing a unit of drug can vary a great deal. Relatively simple drugs, like aspirin, can be very cheap to manufacture, while the complex biological drugs, like Herceptin, can be relatively expensive to manufacture. And the costs don’t stop there. What patients typically refer to as a “drug”, is known as a “drug product”, that is the tablet or injection that patients receive. Before a drug is made into a tablet or injection, it’s known as API (active pharmaceutical ingredient) and is typically a powder of some sort. Since you can’t just give a patient a bottle of powder to take, you need to turn into into a drug product first and that can be either inexpensive (making a tablet) or very expensive (making an inhaler for an asthma medication).

BG-12 (Dimethyl fumarate)
OK, so now that we have all of the industry jargon out of the way, let’s look at an actual example using (very) rough numbers. Biogen Idec has a new drug in clinical trial for multiple sclerosis known as BG-12. BG-12 is very unique in that it is an incredibly simple and inexpensive drug. BG-12 is dimethyl fumarate, a chemical that is manufactured on a huge scale in ton quantities. To give you an example, here is a Chinese chemical supplier who is offering dimethyl fumarate for sale for $1 – $50 per metric ton, but you have to buy at least 2 tonnes and if you need a lot, they can supply up to 2000 metric tons per year. Supply is obviously not an issue here.
If BG-12 is approved by the FDA, patients with multiple sclerosis would receive 240 mg of the drug 2 or 3 times a day (based on clinical trial data). Using rough approximations, let’s calculate how much it would cost to treat one MS patient for a year using BG-12.
Cost of dimethyl fumarate: $25/ton (let’s choose the average cost)
Dose of BG-12 per day: 750 mg (let’s go with the high dose)
Total dose of BG-12 per patient per year: 750 mg * 365 days = 274 g
Total cost of BG-12 per patient per year: $25 * (274g /1,000,000 g) = $0.007
Based on the commodity price of dimethyl fumarate, it would cost less than 1 cent to treat an MS patient with BG-12 for one year. Of course you need to turn the dimethyl fumarate into tablets and put them in a bottle, so let’s assume all those added costs are $0.05/tablet. I honestly have no idea how much it actually costs to make tablets on an industrial scale, but it can’t be much considering you can buy a bottle of 100 generic Tylenol tablets at Target for about $3 or $0.03/tablet all in. However, we’ll estimate on the high side and say $0.05/tablet. If we add in the cost of the dimethyl fumarate ($0.007/tablet) and round up, we get $0.06/tablet or $65.70 to treat an MS patient for a year with BG-12. Not very expensive!
Now we know the variable cost to produce a year’s treatment of BG-12, but how much will it sell for? Well, BG-12 looks like it will be a very effective drug without a lot of side-effects, so Biogen certainly isn’t going to price it less than treatments that are currently on the market. If we take a look at this article from Pharmalot, we can see that the annual cost to treat an MS patient with current drugs ranges from approximately $36,000 to $48,000 per year. Let’s assume that Biogen will match the highest price therapy (Gilenya).
Contribution margin = revenue – variable cost
= $48,000 – $67.50
= $47,932.50 or a 99.85% contribution margin
Now before you start screaming about greedy pharmaceutical companies, remember, this is just the contribution margin. Current estimates put the cost of developing a new drug somewhere between $500M and $2B dollars. Biogen will have to treat a large number of patients for many years to recoup their development costs and begin making a profit on BG-12 (for example: treating 1000 patients with BG-12 for 5 years would pay off $1B in development costs). This is why it’s estimated that only 1 in 3 drugs that hit the market actually make a profit for the company; you may have an incredible contribution margin on a drug, but if all of the margin goes into paying off the fixed costs that went into development, you may end up not making any profit at all.
The other lesson from this exercise is now you can see why the cost of generic drugs is so low. A generic pharmaceutical company incurs only a fraction of development costs that an R&D-based company does and thus can charge a much lower price (typically 80-90% lower), receive a much smaller contribution margin and still make a profit.
Update: After some delay, the FDA has promised to make a decision on BG-12 (brand name: Tecfidera) on March 28th, 2013.
Vivus’ new obesity drug: Will it get scooped by generics before it even hits the market?
Posted: July 2, 2012 Filed under: Latest news 1 CommentThis past Wednesday, the FDA approved Belviq, the recently renamed obesity drug from Arena. It wasn’t a huge surprise since the FDA review panel gave a strong 18-4 vote of support for its approval. Interestingly, it doesn’t appear that the REMS requirement for Belviq is all that onerous (something I commented on in my last blog post), so Arena looks like they have a real winner on their hands.
The next obesity drug up for approval is Qnexa from Vivus. I’ve written about Qnexa in the past (here and here) and it’s been a long journey for Vivus with an initial rejection from the FDA, negotiations over new clinical trial data requirements and a refiling of the NDA. However, after all that, it looks like Qnexa will be approved based on the 20-2 vote by the advisory panel. And that’s a good thing! The clinical trial data for Qnexa is actually much more positive than that for Belviq. Almost 50% of patients taking Belviq lost at least 5% of their body weight (average of 5.7% overall), while 50% of patients taking the highest dose of Qnexa lost at least 15% of their body weight (average of 14.4% overall). You’re probably thinking, “Wow! Vivus has the obesity market cornered!!”. Not so fast, Vivus may get scooped by generics before they even sell their first pill.

Unlike Belviq, which is a new chemical entity and covered under numerous patents which prevents other drug companies from making and selling the active ingredient, Qnexa is a combination of two drugs that are already available as generics (phentermine and topiramate). The highest dose of Qnexa contains 15 mg of phentermine and 92 mg of topiramate. A quick internet search reveals you can get a bottle of 100, 15 mg tablet of phentermine for $142 and a bottle of 120, 100 mg tablets of topiramate for $204, a cost per day of slightly over $3 or $90/month. Yikes! That’s some stiff competition for a branded drug where a price of $150-200 for a month of therapy is seen as a being “on the low end”.
However, Qnexa does an advantage that the generic drugs do not: Qnexa is a controlled-release combination of phentermine and topiramate (this formulation is no doubt patented). This has the benefit that patients can take the drug less frequently and the levels of the drug in the body are much more stable since the dose is slowly release over a period of time. When treating obesity, where food craving and appetite can vary over the course of a day, this is significant advantage. However, it remains to be seen if a physician could simply have the patient split the dose of generic drugs, take it twice a day and see similar results as the sustained-release Qnexa. I have no doubt that physicians will give it a try (or are already trying it).
Two factors will determine the impact of generic phentermine and topiramate on Qnexa’s sales: the price of Qnexa and how insurance companies respond to it. If Qnexa hits the market at a modest premium, say $100 – $120/month, I predict that insurance companies won’t balk at it. They won’t like it, but they’ll also judge the difference in price as too small to devote resources to controlling. However, if the price goes much higher, say $150+, insurance companies will take notice and start to implement some controls that could significant curtail Qnexa sales.
Now insurance companies can’t force a physician to prescribe generic phentermine and topiramate instead of Qnexa, since neither is approved for use in weight loss (physicians on the other hand, are free to prescribe drugs for off-label use). However, insurance companies have the ability to heavily incentivise the use of particular drugs through things like co-pays, step-edits and prior authorizations. The really big risk for Qnexa is the co-pay. Insurance companies typically have “tiers” for their prescription drug coverage that look something like this:
Tier 1: Generic drug: $10 co-pay
Tier 2: Preferred branded drug: $40 co-pay
Tier 3: Non-preferred branded drug: $65 co-pay
Tier 4: Specialty drug: 20% co-insurance
If we imagine a scenario where Qnexa is priced at $120/month (which is way lower than what I think they’ll price it at), insurance companies won’t complain too much about the cost and may choose to put it on tier 2. However, patients will see a significant difference in out-of-pocket expense for Qnexa vs. generic phentermine and topiramate. If a doctor prescribes Qnexa, the patient would pay around $40/month for the prescription. If a doctor prescribes generic phentermine and topiramate, the patient only pays $20 per month ($10 for each prescription). Over a year, that’s a difference of $240, not a lot, but enough to provide some incentive to take the generics. If Qnexa gets price higher than $150, insurance companies may put it on tier 3, where patients are paying $45/month more than the generics for a difference of $540/year. That is a significant amount of money in most people’s books and enough to get patients to ask their physician to prescribe the generic combination.
Either way you cut it, Qnexa, despite have a clear clinical benefit for obese patients, may have a hard time reaching the multi-billion dollar a year sales estimates that have been floating around. When the FDA makes its final call on Qnexa on July 20th, keep an eye out for the price Vivus settles on because it will have a huge impact on how successful (or unsuccessful) the drug becomes.
If the FDA approves the new obesity drugs, will REMS crash the party?
Posted: June 14, 2012 Filed under: Latest news Leave a commentWow! A lot has happened since my last blog post. Two of the three new obesity drugs up for approval (Qnexa from Vivus and Lorquess from Arena) both received positive responses from their respective FDA advisory panels despite all of the pessimism from outside observers. Of course, both drugs have yet to be officially approved by the FDA and they could still be rejected, just ask Intermune.
However, I refuse to be one of the pessimists and I do believe that at least the one of the two drugs will get the FDA’s stamp of approval, if not both. However, before everyone breaks out the champagne and starts celebrating, we need to talk about something called REMS.

REMS stands for Risk Evaluation and Mitigation Strategy and was brought into effect through the 2007 Food and Drug Administration Amendments Act. It was introduced to allow the approval of drugs that have a both an obvious benefit, but also substantial risks. Think of it as a finger that tips the risk-benefit scale more to the benefit side. REMS provides an additional level of control over drugs that otherwise couldn’t be approved for unrestricted use in the general population. If you’re interested in a more detailed overview of REMS, there are some great summaries here and here.
I’m sure you’re wondering what these controls look like. Well, they come in a number of different flavors. Here is a list of typical REMS requirements, starting from the least onerous:
- Medication guide
- Communication plan
- ETASU (Elements to assure safe use)
- Implementation system
The most basic REMS is a medication guide. The guide provides additional information for the prescriber so that they are completely informed of the risks and benefits of the drug and can fully inform the patient taking the drug. It lays out the concerns around adverse events and procedures the prescriber can follow to minimize those risks. The manufacturer provides a draft guide to the FDA, who, if satisfied it contains all the necessary information, approves it.
Pretty simple so far, right? Medication guides are the most common type of REMS and account for around 2/3 of all the drugs that have REMS requirements. The other 1/3 aren’t so lucky, some of those have ETASU requirements.
The types of controls within an ETASU can also vary. They can be as simple as providing formal training for the prescriber so that he or she understands how the drug should be administered, how to educate the patient and how to recognize and report any adverse events. However, on the other end of the spectrum, ETASU controls can also limit who can prescribe the drug, which pharmacies can fill the prescriptions and where the drug can be administered. Where the problem comes in is that you can’t just have these controls, you need mechanisms in place to make sure they are being followed. This requires pharmacists, nurses, physicians and hospitals to take on an incredible administrative burden and a burden that they don’t get reimbursed for. That is how REMS can really put the squeeze on a new drug. If you are a physician who has the choice of prescribing a drug where all you do is write a script versus a drug where your staff has to spend an hour filling out forms just so that the patient can drive across town to the one pharmacy that stocks it, which one would you prescribe? Even if a drug with an onerous REMS is the only drug available to treat a condition, how often will physicians think “I would love to use this drug, but I can justify the burden on me and my staff?”
Both Qnexa and Lorquess have the potential to be approved with a REMS requirement. Qnexa contains topiramate, which is known to increase the risk of cleft palate in mothers who take the drug while pregnant. REMS seems like a great way to keep Qnexa out of the hands of pregnant women, similar to Revlimid (which has an incredibly onerous REMS). Lorquess has the FDA concerned about a possible increased risk of cardiovascular events and REMS would be a great way to collect patient data (through a registry) to determine if that risk is meaningful.
If either of these drugs ends up with strict REMS requirements, you can expect all those financial analysts to quickly revise their revenue estimates and the price of the companies’ stock to react accordingly. So save your champagne until the FDA decisions come out on June 27th (Lorquess) and July 20th (Qnexa). You may just end up saving it for New Year’s Eve.
Are small biotechs more productive because they have no other choice?
Posted: March 21, 2012 Filed under: Innovation Leave a commentOver the last few years, much has been written about the poor R&D productivity of the big pharma companies. There is a bit of a controversy as to whether or not small biotechs are truly more productive, but one can’t deny that many of the new drugs being launched didn’t originate from the labs of companies like Pfizer or Sanofi, but rather small, resource-constrained biotechs like Seattle Genetics (Adcetris) or Optimer (Dificid), to give a couple of examples.
Why is this? Many theories have been bantered around such as biotech’s more collaborative culture, the agility of smaller companies or the science vs. financial-focus. I think all of these ideas have merit, but I would like to examine another cause that I think many people overlook: small biotech companies don’t have a lot of other options.

Now before you say “Yeah, I’ve heard this before, it’s do or die at the small biotechs”, hear me out because that’s not quite where I’m going with this.
Let me start with an anecdote: I was working on a diabetes drug at a big pharma company and during one of our meetings the project lead put up a slide that showed how far behind we were compared to the competition. Our lead compound was in pre-clinical testing, while two of our competitors were already in phase II trials. Yikes, we were at least 3-4 years behind. Why was that? It wasn’t because our program was having trouble, but rather because the program had been put on “pause” almost 5 years before while our resources went to other “higher priority” projects. The project lead was understandably quite frustrated as he had been involved in that initial research and was now being asked “can you please do this faster?”
This type of thing goes on all the time at the big pharma companies; with research budgets in the billions of dollars, R&D portfolios are constantly re-evaluated and resources are reallocated. From the top, this makes sense since why would you put $500M into project A, when putting $500M in project B gives you a bigger NPV (at least according to your model)? The problem is, portfolio strategy is not an exact science and when one of those assumptions you made in your forecasting changes next month, your resource allocation can end up looking completely wrong.
Now contrast that with a small biotech company. A handful of scientists and business folks find some promising technology and decided to develop it. They spend months trying to line up financing and when they do, they have a nice pile of cash that has one purpose: develop the idea they started with. Now this isn’t to say that development plans don’t change, because they do, but the thought never crosses their minds “Hey, maybe we should stop working on this compound and try something different, we can always come back to it.” Even when things appear gloomy and failure seems almost certain, there is no real path other than forward. They aren’t competing against another project for resources because there is no other project. There is no opportunity to put things on “pause”. Projects keep going until they either fail or the money runs out.
What is the consequence of this lack of options? Projects that one day seem like a dead-end keep getting funded (for a while at least) and some of those turns out to be great ideas after all. The same project in a big pharma company? It gets shelved and may never see the light of day again.
How can big pharma fix this problem? Well, the answer isn’t that straightforward. As I mentioned, portfolio strategy is not an easy thing to get right and handing over a multi-billion dollar per year R&D budget to scientists to play with isn’t in the realm of possibility either. What we are seeing is a strategy by big pharma companies to cut R&D budgets and use that cash to support academic research and emerging biotechs. Is this the right strategy? Only time will tell.
